微生物组研究走向绝对定量
Jeroen Raes研究组上周在Nature发表文章,使用Flow cytometry估算粪便中的微生物细胞数量,配合16S测序算出的相对丰度,估算出OTU的绝对数量(absolute abundance)。文章很多结论都印证了直接应用相对丰度进行分析时,我们所看到的很多现象是成分数据性质的假象(可以想象,由于相对丰度在每一个样本中相加为1或100,一个OTU相对丰度增加必将引起其他OTU相对丰度减少,所谓的compositional bias)。特别针对于计算两个OTU的相关系数,当OTU分布不均匀时,很容易看到负相关的OTU–而这仅仅是因为它们受到相加为常数的限制而已。另一个典型例子就是主成分分析(PCA),PCA意在保持欧式距离不变的情况下对数据进行变换,但是相对丰度其实不在欧式空间中(可以参考:J. Aitchison, The Statistical Analysis of Compositional Data, 1986.),这就是为什么在微生物组的研究中更多采用生态学的距离(如,Bray-Curtis distance)来计算\(\beta\) -diversity,然后进行基于距离矩阵的分析(PCoA)。
似乎文章的take home message很简单了,微生物组的研究,我们应该使用类似的方法进行绝对定量。可是仔细想一想,文章指出粪便微生物总量的个体差异可以达到10倍之多,这样大的差异,如果某种微生物在个体之间差异很小,转换成绝对数量之后,个体间的差异将受制于微生物总量。
使用Flow cytometry测定的肠道微生物总量的variation有多大?
下载Nature文章中的Supplementary Table,这里并不不需要购买文章阅读权限,其中表6是Flow cytometry的数据。
加载R包,ggplot2主题
library(tidyverse)## ── Attaching packages ─────────────────────────────────────────────────────────── tidyverse 1.2.1 ──## ✔ ggplot2 2.2.1 ✔ purrr 0.2.4
## ✔ tibble 1.3.4 ✔ dplyr 0.7.4
## ✔ tidyr 0.7.2 ✔ stringr 1.2.0
## ✔ readr 1.1.1 ✔ forcats 0.2.0## ── Conflicts ────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
## ✖ dplyr::filter() masks stats::filter()
## ✖ dplyr::lag() masks stats::lag()library(data.table)##
## Attaching package: 'data.table'## The following objects are masked from 'package:dplyr':
##
## between, first, last## The following object is masked from 'package:purrr':
##
## transposelibrary(cowplot)##
## Attaching package: 'cowplot'## The following object is masked from 'package:ggplot2':
##
## ggsavelibrary(readxl)
figtheme <- theme_bw() +
theme(text = element_text(size=10,face='bold'),panel.border = element_rect(colour = "black",size=2),
axis.title.y=element_text(margin=margin(0,15,0,0)),axis.title.x=element_text(margin=margin(15,0,0,0)),
plot.margin = unit(c(1,1,1,1), "cm"),
plot.title = element_text(margin=margin(0,0,15,0)))
theme_set(figtheme)读取表6,并预处理
cell_counts_dat <- read_excel('~/nature24460-s2.xlsx', sheet=6) %>%
mutate(Cohort=gsub('[0-9]+','',Individual)) %>%
mutate(Cell_count_avg=`Average cell count (per gram of frozen feces)`)## Warning in strptime(x, format, tz = tz): unknown timezone 'zone/tz/2017c.
## 1.0/zoneinfo/Asia/Singapore'cell_counts_dat## # A tibble: 321 x 10
## Individual Cohort Day `Health status`
## <chr> <chr> <dbl> <chr>
## 1 SC01 SC 1 Healthy
## 2 SC02 SC 1 Healthy
## 3 SC03 SC 1 Healthy
## 4 SC04 SC 1 Healthy
## 5 SC05 SC 1 Healthy
## 6 SC06 SC 1 Healthy
## 7 SC07 SC 1 Healthy
## 8 SC08 SC 1 Healthy
## 9 SC09 SC 1 Healthy
## 10 SC10 SC 1 Healthy
## # ... with 311 more rows, and 6 more variables: `Average cell count (per
## # gram of fresh feces)` <chr>, `STDEV cell count (per gram of fresh
## # feces)` <chr>, `Average cell count (per gram of frozen feces)` <dbl>,
## # `STDEV cell count (per gram of frozen feces)` <chr>, Enterotype <chr>,
## # Cell_count_avg <dbl>微生物总量差异
- 个体差异
aggregate(data=cell_counts_dat, Cell_count_avg~Cohort, function(x) c(Mean=mean(x), SD=sd(x), CV=sd(x)/mean(x)) )## Cohort Cell_count_avg.Mean Cell_count_avg.SD Cell_count_avg.CV
## 1 DC 9.092967e+10 6.344747e+10 6.977642e-01
## 2 LC 1.249278e+11 5.406697e+10 4.327858e-01
## 3 SC 1.528339e+11 7.018824e+10 4.592451e-01
## 4 VC 1.447904e+11 7.630896e+10 5.270304e-01ggplot(cell_counts_dat, aes(x=Cohort, y=Cell_count_avg)) + geom_boxplot()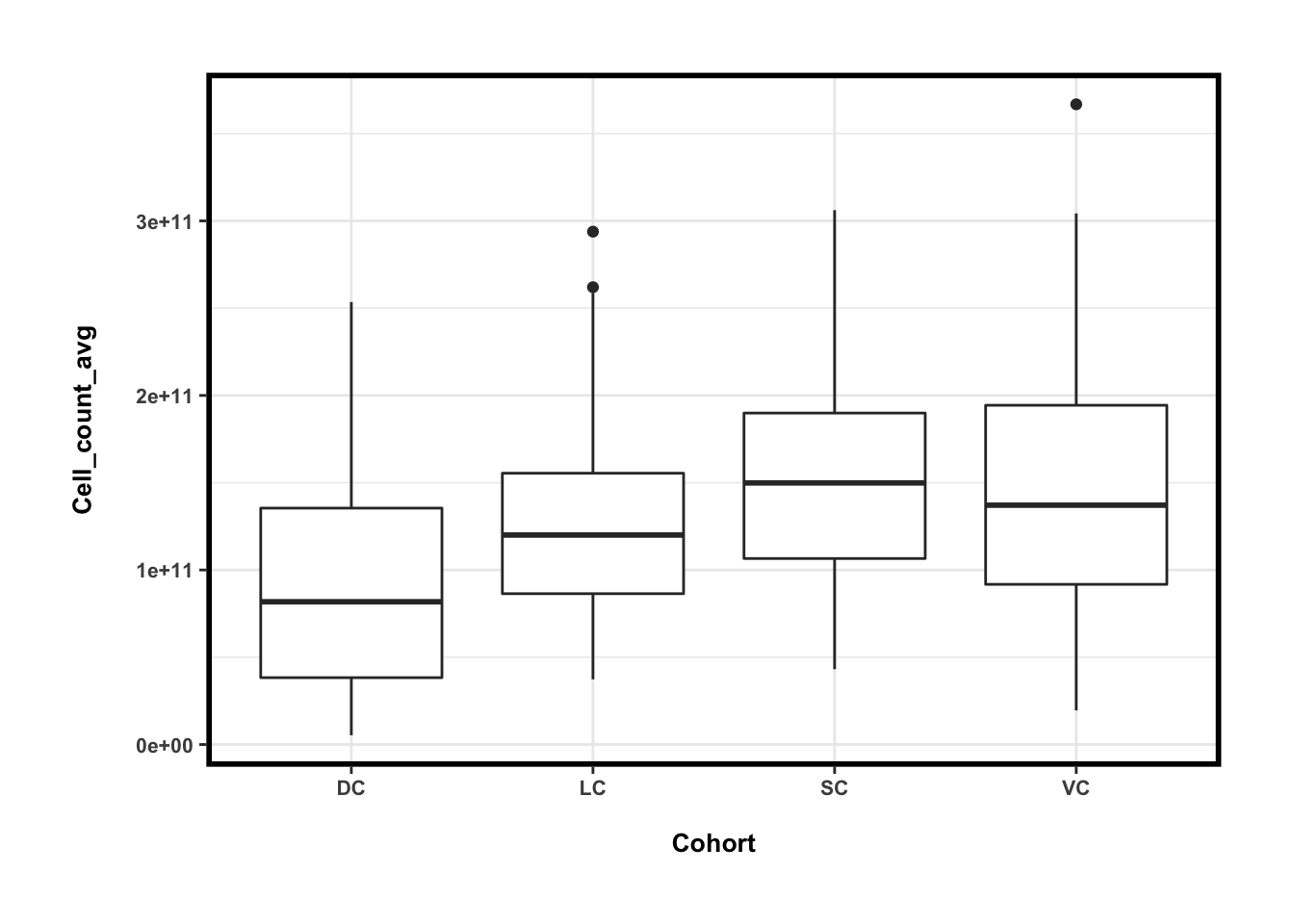
- 个体内差异
aggregate(data=cell_counts_dat[cell_counts_dat$Cohort=='LC',], Cell_count_avg~Individual, function(x) c(Mean=mean(x), SD=sd(x), CV=sd(x)/mean(x)) )## Individual Cell_count_avg.Mean Cell_count_avg.SD Cell_count_avg.CV
## 1 LC01 1.121069e+11 3.062958e+10 2.732175e-01
## 2 LC02 1.057725e+11 1.457955e+10 1.378387e-01
## 3 LC03 6.998669e+10 3.207140e+10 4.582500e-01
## 4 LC04 6.433548e+10 1.478855e+10 2.298661e-01
## 5 LC05 2.198245e+11 2.726579e+10 1.240343e-01
## 6 LC06 2.054927e+11 4.951278e+10 2.409466e-01
## 7 LC07 1.197242e+11 3.438372e+10 2.871911e-01
## 8 LC08 1.162890e+11 3.286685e+10 2.826307e-01
## 9 LC09 1.501435e+11 6.018554e+10 4.008535e-01
## 10 LC10 1.790086e+11 8.293900e+10 4.633241e-01
## 11 LC11 1.565348e+11 4.038210e+10 2.579752e-01
## 12 LC12 1.247153e+11 2.494971e+10 2.000533e-01
## 13 LC13 9.804859e+10 2.625298e+10 2.677548e-01
## 14 LC14 1.037225e+11 4.329933e+10 4.174534e-01
## 15 LC15 1.592359e+11 2.337419e+10 1.467897e-01
## 16 LC16 1.171450e+11 6.207212e+10 5.298741e-01
## 17 LC17 1.215423e+11 3.782074e+10 3.111736e-01
## 18 LC18 1.042144e+11 4.236728e+10 4.065395e-01
## 19 LC19 7.013542e+10 2.326036e+10 3.316492e-01
## 20 LC20 1.465100e+11 2.763866e+10 1.886470e-01ggplot(cell_counts_dat[cell_counts_dat$Cohort=='LC',], aes(x=Day, y=Cell_count_avg)) + geom_line() + facet_wrap(~Individual)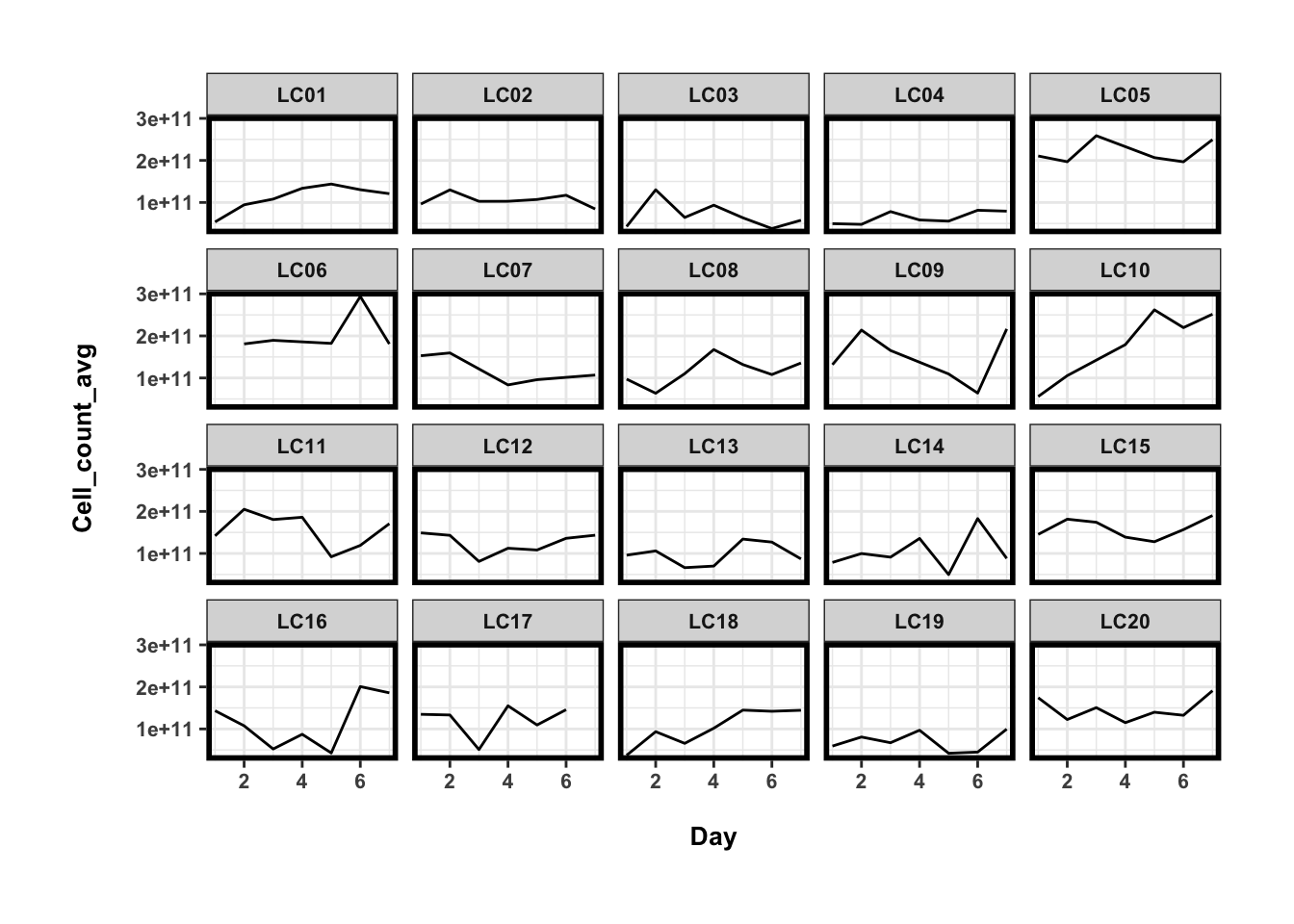
容易看出,疾病组(DC)的微生物总量最低,其他几个健康组微生物总量差异不大,而个人的微生物总量在一周内的变化要小一些(CV~30%),但其实比起个体间差异不是小很多。
人的肠道微生物相对丰度的variation有多大?
下载人类微生物组项目(HMP)数据
我们使用HMPv35的16S的OTU table和元数据表(mapping file)。下载之后解压。
清洗数据,保留肠道数据并去掉整行为0的OTU
metadata <- fread('~/v35_map_uniquebyPSN.txt')
stoolIDs <- as.character((metadata %>% filter(HMPbodysubsite=='Stool'))[,1])
otutab <- fread('~/otu_table_psn_v35.txt',head=TRUE, select=stoolIDs,sep='\t')## Warning in fread("~/otu_table_psn_v35.txt", head = TRUE, select =
## stoolIDs, : Starting data input on line 2 and discarding line 1 because it
## has too few or too many items to be column names or data: # QIIME v1.3.0-
## dev OTU table## Warning in fread("~/otu_table_psn_v35.txt", head = TRUE, select =
## stoolIDs, : Column name '700107040' not found in column name header (case
## sensitive), skipping.## Warning in fread("~/otu_table_psn_v35.txt", head = TRUE, select =
## stoolIDs, : Column name '700114489' not found in column name header (case
## sensitive), skipping.##
Read 44.1% of 45383 rows
Read 45383 rows and 319 (of 4790) columns from 0.409 GB file in 00:00:04## keep samples with counts > 2000
otutab <- otutab %>% select(which(colSums(otutab)> 2000 ))
## we down sample (rarefy) the samples to have 2000 counts in total
otutab_rare <- apply(otutab, 2, function(x) rmultinom(1,size=2000, prob=x))
## remove OTUs not present at all and normalize to proportions
otutab_rel <- apply(otutab_rare[rowSums(otutab_rare)!=0,], 2, function(x)x/sum(x))SD, CV, Mean
SD <- apply(otutab_rel,1,sd)
Mean <- apply(otutab_rel, 1, mean)
tmp <- data.frame(CV=SD/Mean, Mean, SD)
g1 <- ggplot(tmp, aes(y=SD, x=Mean)) +
geom_point(alpha=0.1) +
scale_x_log10() + scale_y_log10() +
annotation_logticks(sides = 'lb')
g2 <- ggplot(tmp, aes(y=CV, x=Mean)) +
geom_point(alpha=0.1) +
scale_x_log10() +
annotation_logticks(sides = 'b')
plot_grid(g1, g2, nrow=1)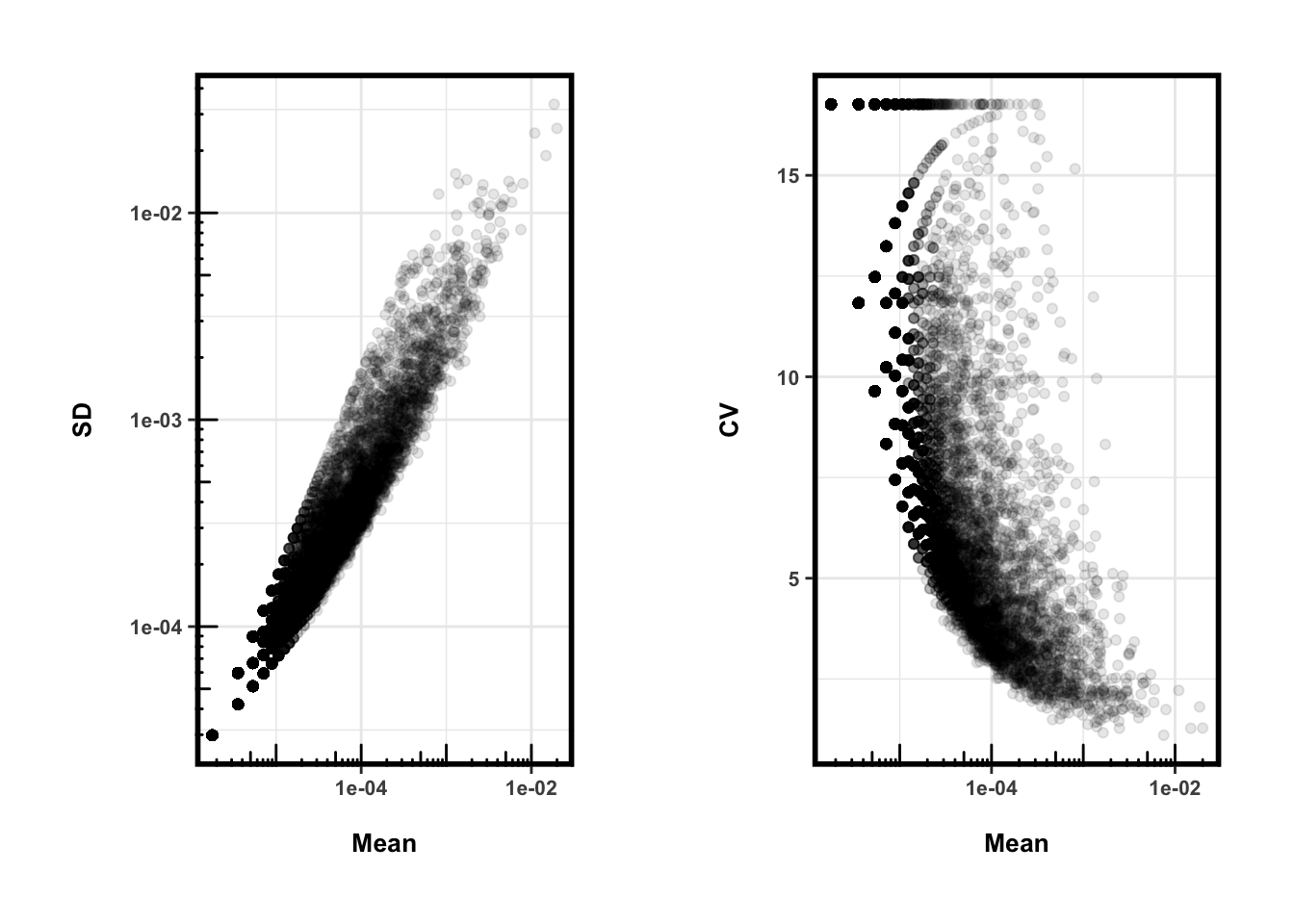
从CV来看,HMP的数据相对丰度其实在个体间差距比较大(特别是低丰度的OTU,可以想象这是跟0的数量多有关),所以可能大多数情况下,我们不会受到总量变化(CV~50%)的影响。Note: 还是需要注意在我们的研究数据中会不会
绝对定量的分析,A thought experiment
正态分布的随机变量(\(M\),\(X\))分别代表微生物总量(\(M\sim|Normal(10^{11},CV_m\times10^{11}|)\)),微生物\(x\)的相对丰度(\(X\sim| Normal(10^{-1},CV_x \times10^{-1})|\)),那么绝对数量\(MX\)会与\(M\)相关吗?
M <- abs(rnorm(1000, 1e11, 0.5e11)) ## CV=0.5
X <- sapply(c(0.1,0.5,1,5,10), function(x)abs(rnorm(1000, 0.1, x*0.1)))
colnames(X) <- c('X_cv0.1','X_cv0.5','X_cv1', 'X_cv5', 'X_cv10')
dat <- melt(X)%>% mutate(MX=value*M) %>% mutate(M=rep(M,5))
dat %>% group_by(Var2) %>% summarise(cor(MX, M, method='spearman'))## # A tibble: 5 x 2
## Var2 `cor(MX, M, method = "spearman")`
## <fctr> <dbl>
## 1 X_cv0.1 0.9745780
## 2 X_cv0.5 0.6241846
## 3 X_cv1 0.4850720
## 4 X_cv5 0.5014045
## 5 X_cv10 0.4692217ggplot(dat,aes(x=M,y=MX)) +
geom_point(alpha=0.4) +
facet_wrap(~Var2, nrow=2, scale='free_y')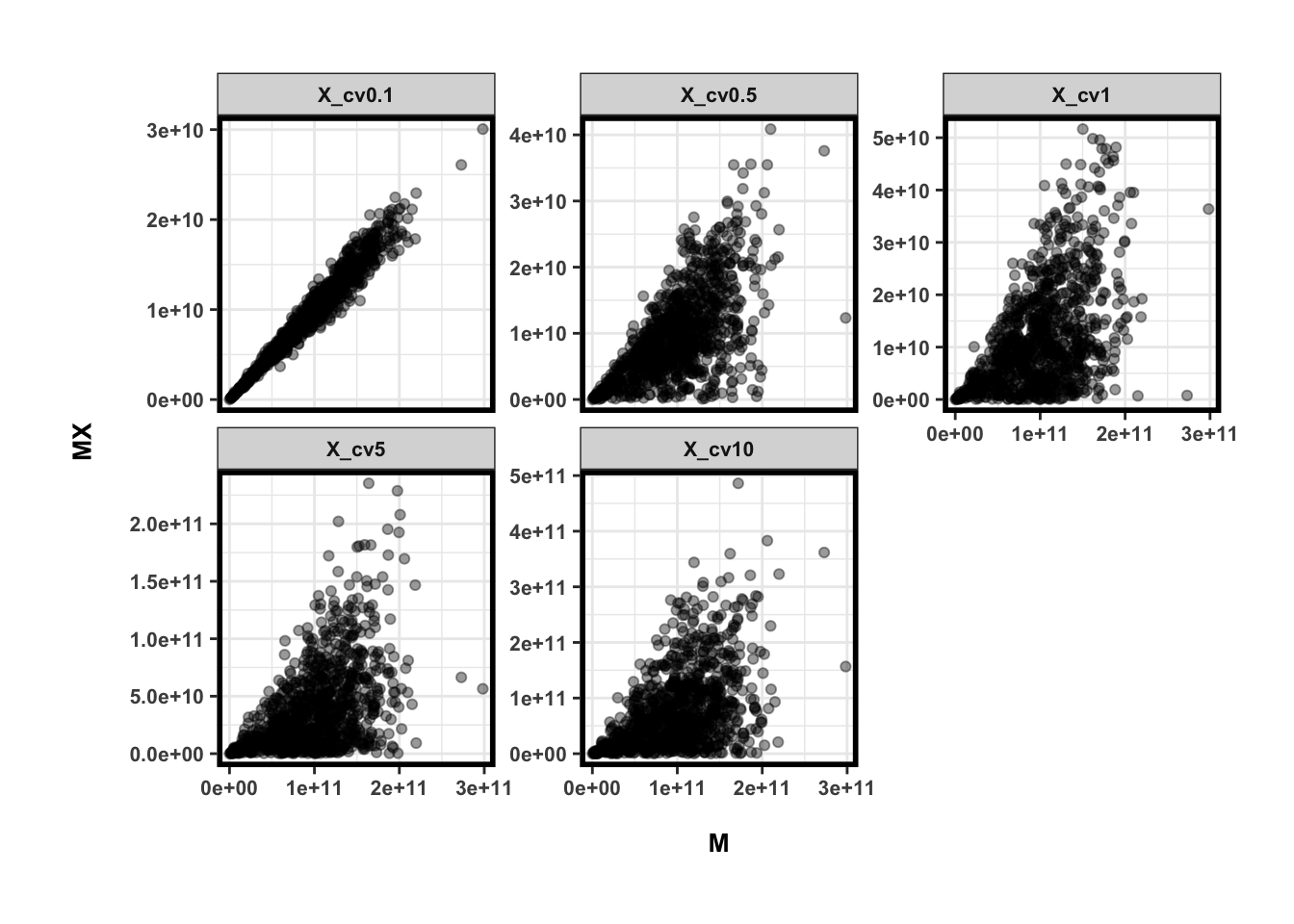
所以不难看出来微生物的绝对数量与微生物总量相关,特别是当相对丰度差异不大的时候。So what?记得correlation有传递性吗?所以如果有两个不相关的微生物,它们的绝对数量会不会因为总量而变得相关呢?我们根据\(X\)的分布生成另一个独立的随机变量\(Y\),让我们来看下他们的correlation是怎样的。
Y <- sapply(c(0.1,0.5,1,5,10), function(x)abs(rnorm(1000, 0.1, x*0.1)))
colnames(Y) <- c('Y_cv0.1','Y_cv0.5','Y_cv1', 'Y_cv5', 'Y_cv10')
dat <- dat %>% mutate(Y=melt(Y)$value) %>% mutate(MY=M*Y)- 毫无关联的\(X\),\(Y\)
dat %>% group_by(Var2) %>% summarise(cor(value, Y, method='spearman'))## # A tibble: 5 x 2
## Var2 `cor(value, Y, method = "spearman")`
## <fctr> <dbl>
## 1 X_cv0.1 0.039534460
## 2 X_cv0.5 0.025382017
## 3 X_cv1 0.039640060
## 4 X_cv5 0.015882484
## 5 X_cv10 0.002462222- 转化成绝对数量后
dat %>% group_by(Var2) %>%
summarise(Spearman_rho=cor(MX, MY, method='spearman'),
p_value=cor.test(MX, MY, method='spearman')$p.value)## # A tibble: 5 x 3
## Var2 Spearman_rho p_value
## <fctr> <dbl> <dbl>
## 1 X_cv0.1 0.9509044 0.000000e+00
## 2 X_cv0.5 0.4455341 0.000000e+00
## 3 X_cv1 0.3194627 0.000000e+00
## 4 X_cv5 0.2866312 2.842296e-20
## 5 X_cv10 0.2930657 3.241526e-21- 散点图
ggplot(dat,aes(x=MX,y=MY)) +
geom_point(alpha=0.4) +
facet_wrap(~Var2, nrow=2, scale='free')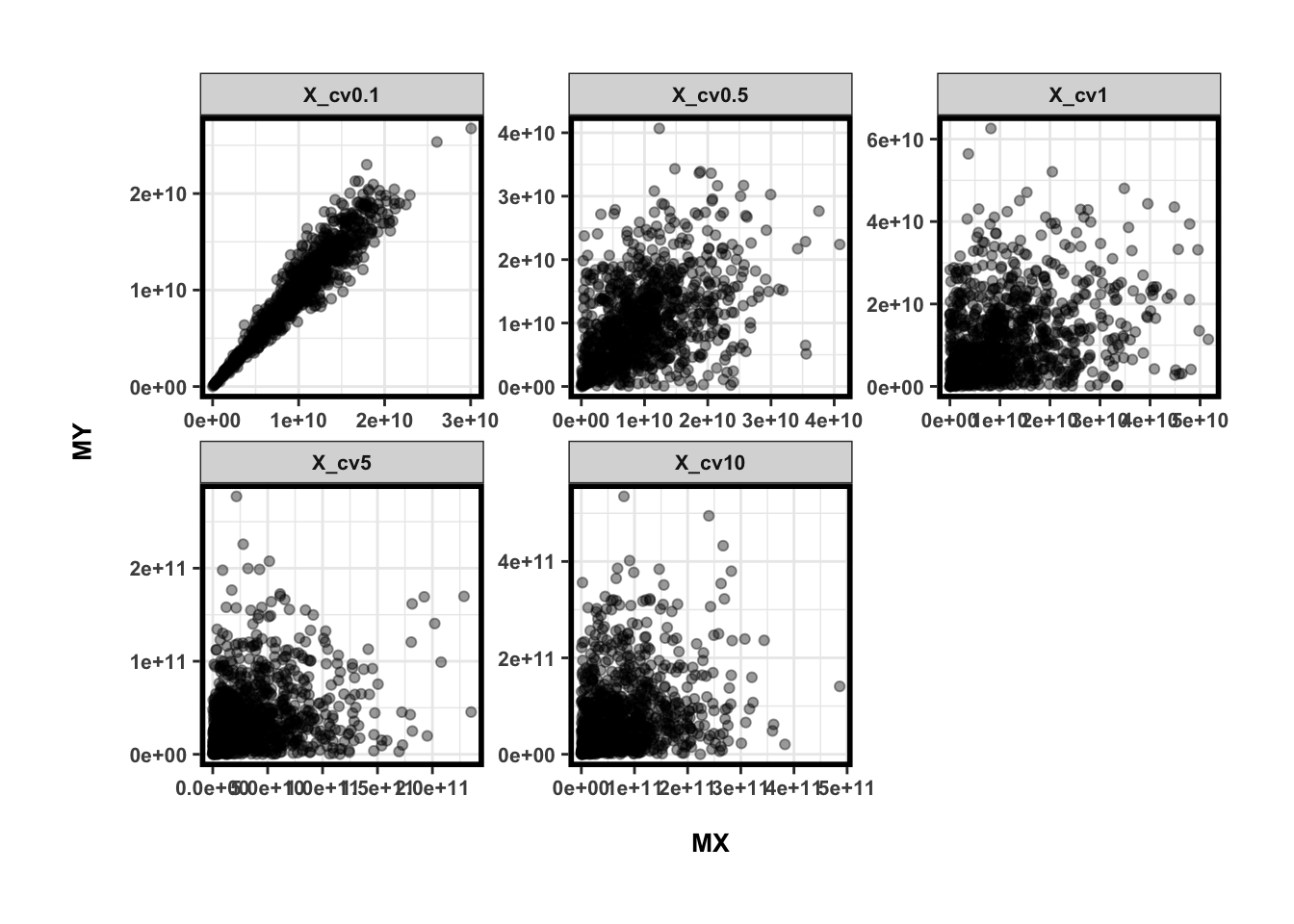
相关性?为什么相关?
所以在以上的实验当中,不难看出即使不相关的两个OTU,因为个体差异不大,转化为绝对数量之后很有可能呈现相关性,而这样的相关性跟生态意义毫无关系,只是因为latent variable微生物总量而已(Nature文章的Figure 3)。
延展开来,当你发现某些微生物的绝对数量和某种表征相关的时候,也需要考虑一下,它们是否只是因为微生物总量的不同呢(Nature文章的Figure 4)?