Data downloading and preprocessing
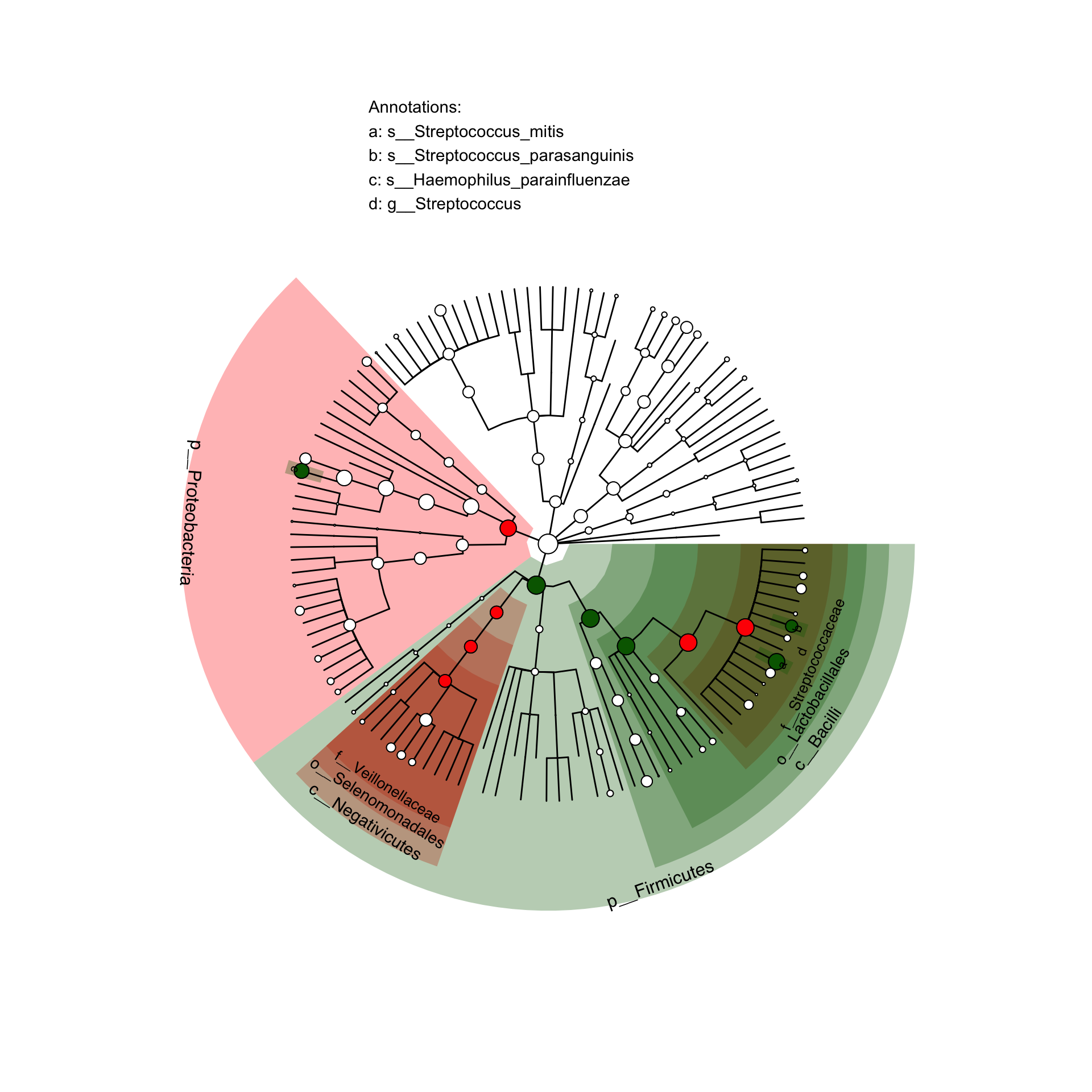
Now let’s try to generate a figure like this:

Sample processed by MetaPhlAn:
https://bitbucket.org/nsegata/metaphlan/wiki/profiled_samples.tar.bz2
Script to merge samples:
Merge files into a single table
python merge_metaphlan_tables.py profiled_samples/*.txt > profiled_samples/merged_abundance_table.txtNow switch to R
Load data and library
df <- read.table("~/Downloads/profiled_samples/merged_abundance_table.txt", head=TRUE, stringsAsFactors = FALSE)
df <- df[,-ncol(df)]
## Use row means as a proxy for node size
dat <- data.frame(V1=df[,1], V2=rowMeans(df[,-1]), stringsAsFactors = FALSE)
library(microbiomeViz)## Warning: replacing previous import 'ape::rotate' by 'ggtree::rotate' when
## loading 'microbiomeViz'Parse data and create a backbone
tr <- parseMetaphlanTSV(dat, node.size.offset=2, node.size.scale=0.8)
p <- tree.backbone(tr, size=0.5)
p
Hard-coded lefse results
lefse_lists = data.frame(node=c('s__Haemophilus_parainfluenzae','p__Proteobacteria',
'f__Veillonellaceae','o__Selenomonadales',
'c__Negativicutes', 's__Streptococcus_parasanguinis',
'p__Firmicutes','f__Streptococcaceae',
'g__Streptococcus','o__Lactobacillales',
'c__Bacilli','s__Streptococcus_mitis'),
color=c(rep('darkgreen',6), rep('red','6')),
stringsAsFactors = FALSE
)Annotate the tree
p <- clade.anno(p, lefse_lists, alpha=0.3)
p